Fibrose cística
O que é a fibrose cística?
A fibrose cística é uma doença hereditária que causa danos graves aos pulmões, sistema digestivo e outros órgãos do corpo.
A doença afeta as células que produzem muco, suor e sucos digestivos, fazendo com que as secreções se tornem pegajosas e espessas.
Ao invés de agirem como lubrificantes, as secreções obstruem os tubos, dutos e passagens, especialmente nos pulmões e no pâncreas.
As melhorias no tratamento, no entanto, têm trazido uma melhor qualidade e expectativa de vida quando comparado com poucas décadas atrás. Atualmente, é comum que os pacientes sobrevivam até seus 30 ou 40 anos de idade, com alguns pacientes vivendo até os 50 anos de idade.
Qual a causa da fibrose cística?
A fibrose cística é uma doença genética, o que significa que ela é herdada dos pais. Ela está associada a uma mutação em um gene chamado de Cystic Fibrosis Transmembrane Regulator (CFTR).
Cada criança recebe dois genes CFTR, um do pai e o outro da mãe. Para desenvolver a doença, ela precisa que ambos os genes tenham a mutação para a fibrose cística. Este tipo de condição é denominada de autossômica recessiva.
Ao receber o gene mutante de apenas um dos pais, a criança não desenvolve a doença. No entanto, ela poderá transmitir o gene mutante para seus filhos.
Qual o risco de um irmão desenvolver a fibrose cística?
A compreensão de como ocorre a transmissão da fibrose cística é fundamental, já que isso determina as chances de futuros membros da família desenvolverem a doença.
Quando duas pessoas normais têm um filho com fibrose cística, isso significa que ambas são portadoras da mutação no gene CFTR. Ou seja, cada uma delas possui um gene CFTR normal e outro mutante.
Cada vez que dois portadores de FC têm um filho, as chances são:
- 25% (1 em 4) de a criança ter fibrose cística
- 50% (1 em 2) de a criança ser portadora, mas sem desenvolver a doença
- 25% (1 em 4) de a criança não ser portadora do gene e não ter FC
Devido a este risco elevado, alguns casais optam, caso queiram novos filhos, por realizar a reprodução assistida.
Isso é melhor explicado na figura abaixo
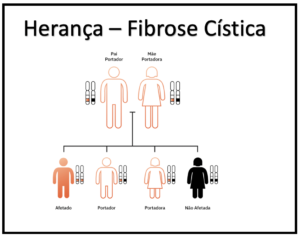
imagem adaptada de https://blog.mendelics.com.br
Diagnóstico da fibrose cística
Teste do pezinho
Todo Recém-nascido no Brasil deve realizar um teste de triagem chamado de teste do pezinho. Este teste busca identificar um conjunto de doenças, incluindo a fibrose cística. Ele deve ser feito mesmo na ausência se sintomas.
O teste do pezinho se baseia na dosagem do tripsinogênio imunorreativo (IRT). A dosagem do IRT é um indicador indireto da doença, pois avalia a integridade da função pancreática, que costuma estar alterada mesmo na ausência de sinais e sintomas.
O teste deve ser realizado no máximo 30 dias após o nascimento. Após este período, o IRT tende a baixar sua concentração e normalizar sua referência no sangue, não devendo mais ser utilizado como exame para triagem. Isso é válido mesmo que a criança seja suspeita de ser portadora de Fibrose Cística.
O IRT tem uma sensibilidade para a fibrose cística ao redor de 95% e especificidade variando entre 32 a 74%, a depender dos níveis de corte estipulados pelos laboratórios para o IRT (1).
Isso significa que 95% dos portadores da doença terão um resultado positivo para o teste (a maioria, mas não todos). No entanto, apenas 32 a 74% dos pacientes que apresentam um teste positivo terão de fato a fibrose cística.
Uma vez que o teste de IRT se mostre positivo, outros exames para confirmação da fibrose cística se fazem necessários, especialmente o teste do suor e o teste genético.
Teste do suor
Para realizar o teste do suor, um produto químico produtor de suor é aplicado a uma pequena área da pele. Em seguida, o suor é coletado para testá-lo e ver se ele está mais salgado do que o habitual.
O teste deve ser realizado na criança com ao menos duas semanas de vida. Ele é indicado para crianças com o teste do IRT positivo, independente dos sinais e sintomas, ou em pacientes sintomáticos, com ou sem um IRT neonatal positivo. Resultados ≥ 60 mmol/L confirmam a fibrose cística.
Teste genético
Existem diferentes formas de mutação do gene CFTR, o que pode implicar no progóstico e no comportamento da doença. Assim, o teste genético pode identificar o tipo específico de mutação de cada paciente.
Além disso, o teste pode ser considerado por irmãos de pacientes com fibrose cística. Estas pessoas têm alto risco de serem portadores da mutação, o que pode ter implicações relacionadas ao planejamento familiar.
A estimativa é de que, no Brasil, haja uma pessoa portadora do gene para cada 25 pessoas. Muitas destas pessoas não têm conhecimento de sua condição de portadoras. Assim, a probabilidade de uma portadora se casar com outra portadora é de aproximadamente 1 em 625. Já a probabilidade de este casal ter um filho com fibrose cística, como vimos acima, é de 1 em 4 para cada filho. Isso leva a uma incidência geral de aproximadamente uma criança com fibrose cística a cada 2.500 nascimentos (1).
No caso de uma pessoa que já conhece sua condição de portadora, a probabilidade de ela casar com outra portadora é de 1 em 25 e de ter um filho com fibrose cística é de 1 em 100 para cada filho.
Devido a este risco aumentado, é possível que se opte pela realização de um teste genético na esposa. Caso ambos sejam portadores, é possível considerar a possibilidade de reprodução assistida, caso assim o desejem.
Quais os sintomas da fibrose cística?
A gravidade da doença e dos sintomas podem variar bastante. Inclusive na mesma pessoa, os sintomas podem piorar ou melhorar significativamente com o passar do tempo.
Algumas pessoas podem levar a infância normalmente e não apresentar sintomas até a adolescência ou a idade adulta.
Quando sintomáticos, uma das principais características da fibrose cística é a quantidade excessiva de sais no suor. Isso pode ser percebido, por exemplo, ao beijar o rosto da criança.
A maioria dos outros sinais e sintomas da fibrose cística estão relacionados à obstrução causada pelo muco nos sistemas respiratório e digestivo.
Sintomas respiratórios
- Tosse persistente com muco espesso (escarro)
- Chiado
- Intolerância ao exercício
- Pneumonias de repetição
- Nariz entupido e com secreção expessa
- sinusite recorrente
Sintomas digestivos
O muco espesso também pode bloquear os ductos pancreáticos, que transportam as enzimas digestivas do pâncreas para o intestino delgado. Sem essas enzimas, o intestino não é capaz de absorver completamente os nutrientes ingeridos, provocando sintomas como:
- Fezes com mau cheiro e gordurosas;
- Baixo ganho de peso e deficit de crescimento;
- Bloqueio intestinal, particularmente em recém-nascidos (íleo meconial);
- Constipação crônica ou grave, podendo inclusive provocar um prolapso retal;
- Pancreatite de repetição.
Complicações respiratórias
Bronquiectasia
A bronquiectasia é uma condição caracterizada pelo alargamento anormal e cicatrização das vias aéreas (tubos brônquicos). Ela leva a uma maior resistência ao fluxo de ar através das vias aéreas. A fibrose cística é uma das principais causas de bronquiectasia.
Infecções crônicas
O muco espesso nos pulmões e seios da face fornece um terreno fértil ideal para bactérias e fungos. Desta forma, pessoas com fibrose cística muitas vezes desenvolvem quadros repetitivos de sinusite, bronquite ou pneumonia.
A infecção por bactérias resistentes a antibióticos e de difícil tratamento também é mais comum.
Pólipos nasais
Pessoas com fibrose cística apresentam um quadro de inflamação crônica da mucosa nasal, facilitando a formação de pólipos nasais.
Tosse com sangue (hemoptise).
Quando a bronquiectasia se desenvolve próximo aos vasos sanguíneos nos pulmões, estes vasos podem ser danificados provocando uma tosse crônica com sangue. Isso pode variar desde pequenos e inocentes sangramentos até quadros bem mais graves, com risco à vida.
Pneumotórax
Pessoas com fibrose cística apresentam risco aumentado para o pneumotórax, uma condição na qual o ar vaza dos pulmões para a caixa torácica, levando ao colapso dos pulmões. Esta é uma complicação que geralmente acomete adultos com fibrose cística, que pode colocar a vida em risco.
Insuficiência respiratória
Com o tempo, a fibrose cística pode danificar tanto o tecido pulmonar que ele não funciona mais. A função pulmonar geralmente piora gradualmente e, eventualmente, pode se tornar uma ameaça à vida. A insuficiência respiratória é a causa mais comum de morte.
Complicações digestivas
Deficiências nutricionais
O muco espesso pode bloquear os tubos que transportam as enzimas digestivas do pâncreas para os intestinos. Sem essas enzimas, o corpo não consegue absorver proteínas, gorduras ou vitaminas lipossolúveis, podendo levar a deficiências nutricionais.
A deficiência nutricional pode levar a complicações como atraso no crescimento, perda de peso, inflamação do pâncreas.
Diabetes
A fibrose cística pode comprometer a capacidade de o pâncreas produzir insulina, o que aumento o risco para diabetes. Cerca de 20% dos adolescentes e 40% a 50% dos adultos com Fibrose cística desenvolvem diabetes.
Doença hepática
Os ductos biliares, responsáveis por transportar a bile do fígado e da vesícula biliar para o intestino delgado, pode ficar bloqueado e inflamado em decorrência da fibrose cística. Isso pode levar a problemas hepáticos, como icterícia, doença hepática gordurosa, cirrose hepática e formação de pedra na vesícula.
Obstrução intestinal
O bloqueio intestinal pode acontecer em pessoas com fibrose cística em todas as idades.
Tratamento da fibrose cística
Nenhuma intervenção disponível atualmente é capaz de prover a cura para a fibrose cística. No entanto, o tratamento pode ajudar no alívio dos sintomas, a reduzir as complicações e a melhorar a qualidade de vida.
Os objetivos específicos do tratamento incluem:
- Prevenir e controlar de infecções pulmonares;
- Remover e soltar o muco dos pulmões;
- Tratar e prevenir a obstrução intestinal;
- Fornecer nutrição adequada.
Tratamento medicamentoso
Dependendo do tipo de mutação genética do paciente, medicamentos moduladores do gene CFTR podem ajudar a melhorar a função da proteína CFTR defeituosa. Eles podem melhorar a função pulmonar e o peso e reduzir a quantidade de sal no suor.
Outros medicamentos são usados para o tratamento dos sintomas e prevenção das complicações da fibrose cística, incluindo:
- Antibióticos para tratar e prevenir infecções pulmonares.
- Medicamentos anti-inflamatórios, para diminuir a inflamação das vias aéreas.
- Mucolíticos, para diluir o muco e permitir que ele seja expelido junto com a toss
- Medicamentos broncodilatadores, para relaxar a musculatura brônquica e manter as vias aéreas abertas.
- Enzimas pancreáticas orais para ajudar o trato digestivo a absorver nutrientes.
- Laxantes, para amolecer as fezes e prevenir a constipação e a obstrução intestinal.
- Antiácidos, para ajudar as enzimas pancreáticas a funcionar melhor.
- Medicamentos específicos para diabetes ou doença hepática, quando apropriado.
Reabilitação e suporte ventilatório
Diferentes técnicas de fisioterapia respitatória podem ser utilizadas para mobilizar e ajudar a eliminar o muco.
Além disso, a fisioterapia deve buscar a melhora ou, ao menos, a preservação da função pulmonar.
Quando a função pulmonar estiver gravemente comprometida, diferentes formas de oxigênioterapia podem ser consideradas. Em alguns casos, como durante o curso de uma infecção respiratória, a oxigenioterapia pode ser uma forma de suporte temporário. Em outras condições, a oxigenioterapia pode ser um tratamento permanente.
A ventilação não invasiva é uma forma de fornecer ar sobre pressão, usada geralmente durante o sono. Ela pode ser feita por meio de uma máscara nasal ou bucal e é muitas vezes usada em combinação com a oxigenioterapia.
O objetivo da oxigenioterapia ou da ventilação invasiva é reduzir o esforço respiratório e aumentar a troca de ar nos pulmões.
Suporte nutricional
A fibrose cística interfere na digestão, além de comprometer a absorção dos nutrientes. Por conta disso, as necessidades energéticas destes pacientes são estimadas em 1 ½ a 2 vezes as necessidades das pessoas sem fibrose cística.
Idealmente, mulheres com fibrose cística devem manter um Índice de Massa Corporal (IMC) de pelo menos 22. Já os homens devem manter um IMC de pelo menos 23. Para pessoas com menos de 21 anos, o IMC deve ser igual ou superior ao percentil 50 no gráfico de crescimento.
Quando o IMC se encontra abaixo destes valores, a dieta deve ainda oferecer cerca de 500 calorias extras ao dia, até que se atinja os valores alvos de IMC.
Para prover este balanço energético, Uma dieta rica em calorias e gordura geralmente recomendada (1). A gordura deve responder por cerca de 40% do total de calorias consumidas.
Quando o consumo calórico indicado estiver sendo muito difícil com a alimentação regular, poderá ser considerado o uso de suplementos alimentares de alto teor calórico. No entanto, estes suplementos devem ter por objetivo apenas complementar, e não substituir a alimentação regular.
Finalmente, quando todas estas estratégias estiverem sendo insuficientes e o aparelho digestivo estiver mais seriamente comprometido, poderá ser considerado o uso de uma sonda nasogástrica ou nasoenteral. Dependendo do caso, a sonda pode ser uma terapia provisória ou definitiva.
Tratamento cirúrgico
As opções para certas condições causadas pela fibrose cística incluem:
Cirurgia nasal
A cirurgia nasal pode ser indicada para a remoção de pólipos nasais, quando estes estiverem obstruindo a respiração. A cirurgia de seio pode ser indicada para o tratamento da sinusite recorrente ou crônica.
Cirurgia do intestino
O tratamento cirúrgico pode ser indicado no caso de uma obstrução intestinal ou de uma intussuscepção – condição em que um segmento do intestino se desloca para dentro de uma seção adjacente do intestino.
Transplante pulmonar
Quando o funcionamento dos pulmões se encontra seriamente comprometido, o transplante de pulmão pode ser uma opção.
A fibrose cística não recorre em pulmões transplantados. No entanto, outras complicações associadas à fibrose cística – como sinusite, diabetes, doenças do pâncreas e osteoporose – ainda podem ocorrer mesmo após um transplante de pulmão.
Transplante hepático
Para doença hepática grave relacionada à fibrose cística, como cirrose, o transplante hepático pode ser uma opção. Em algumas pessoas, um transplante de fígado pode ser combinado com transplantes de pulmão ou pâncreas.