Púrpura Trombocitopênica Trombótica (PTT)
O que é a Púrpura Trombocitopênica Trombótica (PTT)
A Púrpura Trombocitopênica Trombótica (PTT) é uma microangiopatia trombótica auto-imune grave caracterizada pela formação de microtrombos ricos em plaquetas na microcirculação.
Ao “consumir” as plaquetas, esses microtrombos levam a uma redução na contagem de plaquetas (trombocitopenia). Além disso, os microtrombos provocam uma destruição mecânica das células sanguíneas, resultando em anemia hemolítica microangiopática.
Os sintomas estão relacionados principalmente aos sangramentos fáceis e à obstrução ao fluxo sanguíneo, com isquemia tecidual. A maior preocupação está relacionado ao acometimento da circulação sanguínea cerebral.
A doença ocorre com maior frequência em adultos, com pico de incidência após os 40 anos, embora formas congênitas possam afetar crianças.
Fisiopatologia
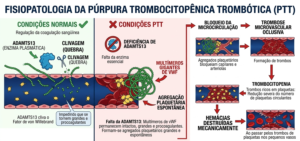
A PTT é uma condição autoimune que leva a uma deficiência da ADAMTS13, uma enzima plasmática essencial para a regulação da coagulação sanguínea.
Em condições normais, a ADAMTS13 cliva (quebra) o fator de von Willebrand à medida que são liberados pelas células endoteliais, impedindo que eles se tornem grandes e procoagulantes.
No paciente com PTT, a falta dessa enzima faz com que se acumulem polímeros do fator de von Willebrand com alto peso molecular (não clivados), o que favorece a coagulação sanguínea.
Os coágulos formados bloqueiam a microcirculação (capilares e arteríolas), resultando em trombose microvascular oclusiva.
A formação desses trombos de forma disseminada consome grande parte das plaquetas circulantes, o que leva a uma redução severa do número de plaquetas (trombocitopenia).
Ao mesmo tempo, as hemácias são destruídas mecanicamente ao passar pelos trombos nos pequenos vasos – levando a uma anemia.
Os trombos fazem com que o fluxo de sangue nas artérias obstruídas seja interrompido. Um dos órgãos mais sensíveis a essa obstrução é o cérebro. Embora nem todos apresentem danos neurológicos, estes são os que mais preocupam. Algumas das queixas que podem estar preentes nesses casos são a confusão mental, dor de cabeça, convulsões ou mesmo coma.
Sinais e sintomas
Os sinais mais comuns da Púrpura Trombocitopênica Trombótica estão relacionados a sangramentos (decorrentes da plaquetopenia), à anemia, sintomas neurológicos (decorrentes da isquemia cerebral) e sintomas decorrentes da isquemia em outros órgãos.
- Plaquetas Baixas e Sangramentos: Púrpura (manchas roxas), equimoses, petéquias (pontos vermelhos) e sangramentos mucosos (nasal, gengival).
- Anemia: Fadiga, fraqueza, palidez e icterícia (pele e olhos amarelados).
- Sintomas Neurológicos: Alterações no estado mental, dor de cabeça (cefaleia), confusão, convulsões e, em casos graves, coma ou derrame (AVC).
- Comprometimento de Órgãos: Febre, dores abdominais, náuseas, vômitos e insuficiência renal.
Púrpura Trombocitopênica imune X trombótica
A Púrpura Trombocitopênica Imune (PTI) e a Púrpura Trombocitopênica Trombótica (PTT) são distúrbios sanguíneos que resultam em baixa contagem de plaquetas (trombocitopenia) e sangramentos frequentes. No entanto, elas possuem mecanismos, gravidade e tratamentos completamente diferentes.
A PTT é uma emergência médica grave, enquanto a PTI é frequentemente uma doença crônica e manejável. Mostramos na tabela abaixo as principais diferenças e semelhanças entre elas.
| Púrpura Trombocitopênica Imune X Trombótica | ||
| Característica | Púrpura Trombocitopênica Imune (PTI) | Púrpura Trombocitopênica Trombótica (PTT) |
| Mecanismo | Destruição de plaquetas por anticorpos (Autoimune). | Deficiência da enzima ADAMTS13 |
| Plaquetas | Baixas (Trombocitopenia). | Muito baixas (Trombocitopenia). |
| Gravidade | Geralmente crônica, com baixo risco de morte. | Emergência médica grave. |
| Hemólise | Incomum | Sim (Anemia Hemolítica Microangiopática). |
| Sintomas Principais | Hematomas, petéquias, sangramento gengival. | Febre, disfunção neurológica (AVC, confusão), insuficiência renal. |
| Tratamento | Imunossupressores (corticoide), remoção do baço. | Plasmaférese urgente (troca plasmática). |
Diagnóstico
O diagnóstico da PTT é essencialmente clínico-laboratorial e deve ser feito de forma imediata, devido aos prejuízos graves associados ao atraso no início do tratamento.
No paciente com quadro clínico característico, o primeiro passo é a confirmação laboratorial de uma anemia hemolítica microangiopática, o que envolve três achados:
- Trombocitopenia (plaquetas baixas)
- Anemia (hemoglobina baixa)
- Presença de esquizócitos no esfregaço
Os esquizócitos são fragmentos de hemácias que foram destruídas ou danificadas enquanto circulavam pelos vasos sanguíneos. Eles indicam que houve um dano mecânico às células, sendo um achado característico das anemias hemolíticas microangiopáticas.
Diferenciação com outras anemias hemolíticas microangiopáticas
Os principais diagnósticos diverenciais nesses casos são a Coagulação Intravascular Diceminada (CIVD) e a Síndrome Hemolítico-Urêmica (SHU).
Do ponto de vista clínico, podemos considerar que:
- PTT: Geralmente idiopática ou associada a doenças autoimunes, sem antecedente infeccioso evidente.
- SHU: geralmente precedida por um quadro de diarreia sanguinolenta.
- CIVD: presença de uma doença de base, como sepse, câncer, choque ou complicações obstétricas.
Na tabela abaixo, mostramos as principais diferenças laboratoriais entre essas condiçòes:
| Doença de Base | Síndrome Hemolítico Urêmica (SHU) | Púrpura Trombocitopênica Trombótica (PTT) | Coagulação Intravascular Disseminada (CIVD) |
| Contagem de Plaquetas | Reduzida | Reduzida | Reduzida |
| Anemia Hemolítica Microangiopática | Presente | Presente | Presente |
| TP (INR) | Normal | Normal | Aumentado |
| TTPA | Normal | Normal | Aumentado |
| Fibrinogênio | Normal | Normal | Reduzido |
| D-dímero | Normal | Normal | Aumentado |
| Atividade do ADMATS13 | Normal | Reduzido |
O primeiro passo na investigação desses pacientes é avaliar se o paciente apresenta anormalidades de coagulação (pela testagem laboratorial do TP e o TTPA). Quando esses exames estão alterados, devemos pensar em CIVD.
Por outro lado, exames de coagulação normais nos leva a pensar em Púrpura Trombocitopênica Trombótica ou Síndrome Hemolítico Urêmica. Do ponto de vista clínico, a PTT pode levar a um quadro neurológico mais evidente, mas com disfunção renal mais leve. Já na SHU a insuficiência renal tende a ser bem mais grave.
Além disso, a diferenciação pode ser feita por meio da avaliação da Atividade do ADMATS13. Níveis de atividade inferiores a 10% indicam deficiência grave, confirmando Púrpura Trombocitopênica Trombótica. Quando a atividade enzimática está normal, fica caracterizada a Síndrome Hemolítico Urêmica.
Tratamento
A PTT é uma emergência hematológica. O paciente deve ser internado assim que for feita a suspeita diagnóstica, preferencialmente em unidade com suporte intensivo, e o tratamento iniciado antes mesmo da confirmação laboratorial.
Plasmaférese
O tratamento com plasmaférese é a base do tratamento, devendo ser iniciado sem aguardar o resultado da atividade de ADAMTS13.
Nesse procedimento, o sangue é retirado da pessoa e colocado em um aparelho que separa as células sanguíneas da parte líquida do sangue (plasma). O plasma, que contém os anticorpos causadores da doença, é descartado. Já as células sanguíneas são devolvidas à pessoa, juntamente com plasma fresco obtido de um banco de sangue.
Dessa forma, a plasmaférese remove anticorpos contra ADAMTS13 e repõe a enzima funcional. Isso reduz formação de microtrombos e a hemólise e melhora rapidamente os níveis de plaquetas. Ela deve ser feita diariamente até a normalização das plaquetas e a resolução da hemólise.
Corticoesteroides
O corticoesteroide é um tratamento de primeira linha, Juntamente com a plasmaférese. Os corticoides reduzem a resposta autoimune e diminuem produção de anticorpos.
Geralmente ele é feito com metilprednisolona por via intravenosa, durante 1 a 3 dias, para controle rápido da inflamação.
Imunoterapia
Nos casos de doença grave, recorrente ou refratária ao tratamento, a imunoerapia com Rituximabe deve ser considerada. O Rituximabe atua reduzindo produção de autoanticorpos.
Transfusão
A transfusão sanguínea poderá ser indicada em caso de anemia sintomática.
Por outro lado, a transfusão de plaquetas deve ser evitada, uma vez que ela pode piorar a formação de trombos. Ela só deve ser feita em caso de sangramento grave com risco de vida.
Monitoramento
Os níveis de plaquetas geralmente começam a aumentar em 2 a 5 dias após o início da plasmaférese, com normalização em 7 a 14 dias.
A resposta ao tratamento deve ser monitorada diariamente pela contagem de plaquetas, níveis da enzima ADAMTS13 e níveis de DHL (indicativo da hemólise).
Aplasmaférese deve ser repetida diariamente, até a normalização das plaquetas e da hemólise, o que significa 7 a 14 sessões em média.
Prognóstico
Sem tratamento, a mortalidade por PTT chaga a aproximadamente 90% dos casos. Com o tratamento, a sobrevida é de aproximadamente 90%.
Entre os sobreviventes, o risco de recorrência da PTT é alto, podendo chegar a 30% a 50% dos pacientes. Por isso, o acompanhamento hematológico contínuo por meio para contagem de plaquetas ee dosagem do DHL se faz necessário, especialmente no primeiro ano após o evento agudo.
Por fim, é preciso considerar que alguns pacientes poderão ter sequelas permanentes em decorrência do comprometimento cerebral, especialmente relacionados a déficits cognitivos leves, alterações neuropsiquiátricas ou doença renal leve residual.