Talassemia
O que é a Talassemia
A talassemia é um grupo de doenças genéticas caracterizadas pela redução ou ausência na síntese de uma ou mais cadeias de globina da hemoglobina, principalmente as cadeias alfa (α) ou beta (β). Esse defeito leva à formação de hemácias anormais, que são precocemente destruídas. O resultado final é uma anemia crônica, do tipo microcítica e hipocrômica.
Alguns tipos podem levar a uma anemia leve com pouco impacto na saúde, sem necessidade de tratamentos específicos. Outros podem levar a a uma anemia grave, com necessidade de transfusões de sangue frequentes e risco à vida.
As formas leves são muito comuns, sendo que cerca de 7% da população mundial é portadora de algum traço talassêmico. Já as formas mais graves e com necessidade de transfusões regulares são raras. No Brasil, há cerca de 1.000 a 1.300 pessoas com formas graves de talassemia cadastradas, a maioria deles na região Sudeste.
Quais os Tipos de Hemoglobina?
A hemoglobina pode ser classificada em alguns tipos de acordo com os seus constituintes.
Ela é formada pelo grupamento heme (ferro), e pelas cadeias globinas, que podem ser alfa, beta, gama ou delta.
A composição das globinas é que determinam os principais tipos de hemoglobina, incluindo:
- HbA1: formada por duas cadeias alfa e duas cadeias beta e está presente em maior concentração no sangue;
- HbA2: formada por duas cadeias alfa e duas cadeias delta;
- HbF: formada por duas cadeias alfa e duas cadeias gama. Está presente em maior concentração em recém-nascidos, tendo sua concentração diminuída de acordo com o desenvolvimento.
Tipos de Talassemia
Pode ser dividida em dois tipos:
- Talassemia Alfa: deficiência na produção das cadeias alfa;
- Talassema Beta: deficiência na produção das cadeias Beta.
Talassemia Alfa
Quatro genes estão envolvidos na formação da cadeia alfa da hemoglobina, sendo que dois genes são herdados de cada um dos pais.
A gravidade depende do número de mutações genéticas herdadas dos pais. Quanto mais genes mutados, mais grave é a Talassemia.
Assim, a Alfa-talassemia pode ser classificada em três tipos:
- Um ou dois genes afetados (traço talassêmico): O paciente não apresenta sinais ou sintomas ou tem sinais e sintomas leves.
- Três genes afetados (Doença da Hb H). A anemia e os sintomas serão moderados a graves.
- Quatro genes afetados (Síndrome da hidropsia fetal): forma mais grave das síndromes talassêmicas, sendo causa de morte intra-uterina ou morte logo após o nascimento.
Talassemia Beta
Dois genes estão envolvidos na formação da cadeia beta da hemoglobina. Um deles é herdado do pai, o outro da mãe.
Ela pode ser classificada nos seguintes tipos, de acordo com as mutações envolvidas:
- Beta Talassemia Menor (ou Traço Talassêmico): É a forma mais leve, onde a pessoa possui apenas um gene beta alterado. Geralmente, não há sintomas ou apenas anemia muito leve, sendo comum ser diagnosticada apenas em exames de rotina.
- Beta Talassemia Intermediária: tem um espectro clínico variável, com anemia moderada. Os pacientes geralmente não dependem de transfusões sanguíneas frequentes, mas podem precisar delas em certas ocasiões (como infecções ou gravidez).
- Beta Talassemia Maior (Anemia de Cooley): Forma mais severa. Ocorre quando ambos os genes beta estão alterados, impedindo a produção de hemoglobina funcional. Os pacientes necessitam de transfusões de sangue regulares a cada 2-4 semanas desde os primeiros meses de vida.
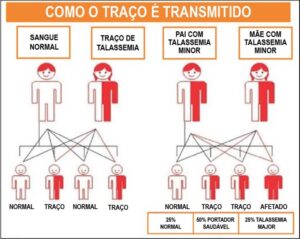
Padrão de Herança na Talassemia
Compreender o padrão de herrança na talassemia é fundamental, especialmente no caso de familias que têm um fiho com talassemia e pretendem ter outros filhos. Devido ao risco elevado de novos casos nas futuras gestaçõe, alguns casais podem optar por técnicas de reprodução assistida.
Padrão de Herança na Talassemia Beta
A talassemia beta é uma condição autossômica recessiva. Assim, quando ambos os pais apresentam um único gene alterado, eles podem ter sintomas leves ou ausentes e muitos não saberão serem portadores, sendo, portanto, considerados com traço da talassemia.
Quando ambos os pais apresentam traço talassêmico, poderão ser observados os seguintes padrões de herança:
- 25% de chance de o filho ter a talassemia maior (doença grave).
- 50% de chance de o filho herdar apenas o traço (portador saudável).
- 25% de chance de o filho ter sangue normal (não afetado).
Quando um casal tem um filho com a forma grave da talassemia, dessa forma, é recomendado que seja feita uma consulta de aconselhamento genético, já que há um risco elevado de que eventuais novos filhos também desenvolvam a doença. Algumas famílias podem optar por técnicas de reprodução assistida nessas situações.
Padrão de Herança na Talassemia Alfa
A produção da proteína alfa-globina é regulada por quatro genes da alfa-globina: dois (um de HBA1 e um de HBA2) em cada cópia do cromossomo 16. A gravidade da condição está correlacionada com o número de genes alterados e, portanto, com a quantidade de cadeias de alfa-globina produzidas.
Um indivíduo herda um cromossomo com dois genes da alfa-globina de cada progenitor. Assim, ele possui um total de quatro genes da alfa-globina (α α/ α α) para produzir a quantidade necessária de cadeias alfa.
A Alfa talassemia está associada a uma delação desses genes – variando da deleção de um gene (- α/ α α) até a deleção dos quatro genes (- – / – -).
- Um ou dois genes afetados (traço talassêmico): O paciente não apresenta sinais ou sintomas ou tem sinais e sintomas leves.
- Três genes afetados (Doença da Hb H). A anemia e os sintomas serão moderados a graves.
- Quatro genes afetados (Síndrome da hidropsia fetal): forma mais grave das síndromes talassêmicas, sendo causa de morte intra-uterina ou morte logo após o nascimento.
Pacientes com traço talassêmico se dividem em dois grupos: alfa plus (α – / α –) ou alfa zero (- – / α α), o que é importante no sentido de aconselhamento genético e na probabilidade de que o casal tenha novos filhos com a doença.
Vamos considerar aqui as seguintes situações:
Situação 1: ambos os pais apresentam mutação alfa zero
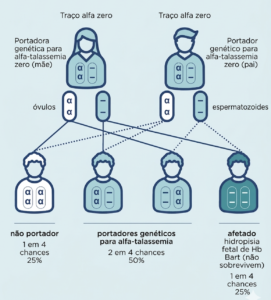
Em um casal no qual ambos são portadores genético do traço de alfa-talassemia zero, em cada gravidez há:
- 25% de probabilidade do filho herdar ambas as cópias da variante recessiva do gene dos pais. Nesse caso, nenhum produto gênico funcional será produzido e a criança terá hidropsia fetal e não sobreviverá.
- 25% de probabilidade do filho herdar ambas as cópias do gene funcional. Assim, ele não terá alfa-talassemia, nem será um portador genético.
- 50% de probabilidade do filho herdar a variante recessiva do gene e a cópia funcional do gene dos pais, sendo um portador genético assintomático de alfa-talassemia, assim como os pais.
Situação 2: um pai é portador do traço alfa zero e o outro do traço alfa plus
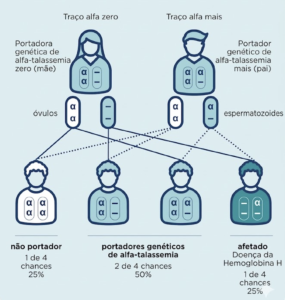
Quando um dos pais é portador do traço de talassemia alfa zero e o outro é portador do traço de talassemia alfa plus, em cada gravidez há:
- 25% de probabilidade do filho herdar ambas as cópias da variante genética recessiva dos pais. Nesse caso, haverá produção reduzida da proteína funcional e a criança terá doença da hemoglobina H.
- 25% de probabilidade de um filho herdar ambas as cópias do gene funcional. Ele não terá talassemia alfa nem será portador genético.
- 50% de probabilidade de um filho herdar a variante genética recessiva e a cópia funcional do gene dos pais, sendo portador genético assintomático da talassemia alfa, assim como qualquer um dos pais.
Quais os sintomas da Talassemia?
Os sinais e sintomas da talassemia variam de acordo com a gravidade da doença (maior, intermediária ou menor/traço) e a idade do paciente. Eles estão relacionados à anemia crônica e à destruição de glóbulos vermelhos.
Recém-nascidos e Bebês (0 a 2 anos)
Os sintomas nessa faixa etária estão relacionados principalmente à Talassemia Maior. Eles geralmente se manifestam após os 6 meses de vida, quando a hemoglobina fetal é substituída pela hemoglobina do adulto (defeituosa), podendo incluir:
- Palidez intensa
- Sonolência e irritabilidade, apatia ou irritação excessiva.
- Fadiga
- Baixo apetite e problemas para se alimentar.
- Icterícia:Pele ou olhos amarelados.
- Aumento do baço (esplenomegalia) e do fígado.
- Crescimento lento
Crianças (2 a 12 anos)
Sintomas nessa faixa etária costumam ser percebidos tanto na Talassemia Maior quanto na intermediária, podendo incluir:
- Anemia grave, com necessidade de transfusões de sangue (na forma maior).
- Deformidades ósseas: Alterações na face e crânio (ossos da face proeminentes, maxilar superior saliente). Isso acontece por conta do aumento e hiperatividade da medula óssea, tentando compensar a falta de sangue.
- Atraso no crescimento e desenvolvimento, com crianças menores e mais magras que a média para a idade.
- Problemas ortodônticos, especialmente com mal alinhamento dentário.
- Sistema imune enfraquecido, com infecções frequentes.
- Barriga aumentada, por conta do aumento do baço.
Adolescentes e Adultos
A Talassemia Minor (Traço Talassêmico) geralmente segue assintomática, embora possa levar a uma leve anemia, com cansaço discreto. Os sintomas são mais evidentes nas Talassemias Maior/Intermediária, podendo incluir:
- Atraso na puberdade.
- Problemas cardíacos decorrentes do acúmulo de ferro (hemossiderose) ou anemia crônica, incluindo arritmias ou insuficiência cardíaca.
- Problemas no fígado e diabetes, também pelo excesso de ferro.
- Icterícia (Pele amarelada).
- Feridas na pele.
- Fadiga importante ou extrema.
Complicações
As possíveis complicações da Talassemia moderada a grave incluem:
Sobrecarga de ferro
Pessoas com talassemia podem ter excesso de ferro em seus corpos, seja pela doença ou por transfusões de sangue frequentes.
O excesso de ferro é tóxico principalmente para o coração, fígado e sistema endócrino, podendo resultar em insuficiência cardíaca (principal causa de morte), cirrose hepática, diabetes e distúrbios hormonais. A terapia de quelação de ferro contínua é necessária para evitar danos graves.
Infecção
Pessoas com talassemia têm um risco aumentado de infecção. Isso é especialmente verdadeiro no caso de remoção do baço.
Deformidades Ósseas
A Talassemia pode fazer com que a medula óssea se expanda e os ossos se alarguem. Isso pode resultar em estrutura óssea anormal, especialmente no rosto e crânio.
A expansão da medula óssea também torna os ossos finos e quebradiços, aumentando o risco para fraturas.
Esplenomegalia (baço aumentado)
O baço é um órgão importante para o combate a infecções e para filtrar material indesejado, incluindo as células sanguíneas velhas ou danificadas.
A Talassemia é frequentemente acompanhada pela destruição de um grande número de glóbulos vermelhos. Isso faz com que o baço aumente e trabalhe mais do que o normal.
Um baço aumentado (esplenomegalia) pode piorar a anemia e reduzir a vida dos glóbulos vermelhos transfundidos. Se o baço ficar muito grande, poderá ser indicada uma cirurgia para removê-lo.
Retardo de Crescimento
A anemia pode retardar o crescimento de uma criança e atrasar a puberdade.
Problemas cardíacos
Insuficiência Cardíaca Congestiva e Arritmia Cardíaca podem estar associados à Talassemia grave.
Diagnóstico
O traço talassêmico é comumente detectado quando o exame direto do sangue periférico de rotina e hemograma completo revelam uma anemia microcítica e alta contagem de eritrócitos.
Já a talassemia moderada a grave costuma se manifestar clinicamente nos primeiros dois anos de vida.
O diagnóstico pode ser confirmado por meio do exame de sangue, quando se observa uma anemia hemolítica microcítica.
Os níveis séricos de bilirrubina, ferro e ferritina também estão aumentados.
Na talassemia beta maior, a anemia é grave, geralmente com hemoglobina ≤ 6 g/dL. A contagem de eritrócitos é elevada em relação à hemoglobina.
O esfregaço sanguíneo é praticamente diagnóstico, mostrando alterações morfológicas características.
Tratamento
Formas leves de traço de talassemia não precisam de tratamento.
Para talassemia moderada a grave, os tratamentos a serem considerados pelo Médico Hematologista podem incluir:
Transfusões de sangue frequentes
Formas mais graves de Talassemia geralmente requerem transfusões de sangue frequentes, em muitos casos a cada poucas semanas.
Com o tempo, as transfusões de sangue causam um acúmulo de ferro no sangue, o que pode danificar o coração, o fígado e outros órgãos.
Terapia de Quelação
Este é um tratamento para remover o excesso de ferro do sangue.
Algumas pessoas com talassemia que não fazem transfusões regulares também podem desenvolver excesso de ferro.
A quelação é feita por meio de medicamentos específicos. Ela é geralmente iniciada quando os níveis de ferritina sérica estiverem > 1.000 ng/mL ou após cerca de 1 a 2 anos de transfusões programadas.
Esplenectomia
A esplenectomia (remoção do baço) pode ser considerada no caso de pacientes com necessidade de transfusões frequentes, na presença de esplenomegalia.
Ela pode diminuir a destruição das hemácias e a necessidade de transfusão.
Transplante de Medula Óssea
Para crianças com talassemia grave, o Transplante de Medula Óssea pode eliminar a necessidade de transfusões de sangue ao longo da vida.
Consequentemente, elimina também a necessidade de medicamentos para controlar a sobrecarga de ferro.
A transfusão envolve infusões de células-tronco de um doador compatível, geralmente um irmão.
Alimentação
A sobrecarga de ferro é uma consequência já esperada com a Talassemia, devido às frequentes transfusões.
Assim, é importante que se evite uma dieta com excesso de ferro. Suplementos que contenham ferro geralmente também devem ser evitados.
Por outro lado, suplementos de ácido fólico e de vitamina B podem ser indicados para ajudar na produção de novos glóbulos vermelhos.
Medidas para evitar infecções
O paciente com talassemia encontra-se mais vulnerável para infecções. Assim, alguns cuidados devem ser considerados para minimizar este risco.
Isso inclui lavar as mãos com maior frequência e evitar o contato com pessoas doentes.
Isso é especialmente importante para aqueles que tiveram que remover o baço.
O calendário vacinal deve incluir a vacina anual contra gripe e vacinas contra meningite, pneumonia e hepatite B.