Feocromocitoma e Paraganglioma
O que é o feocromocitoma e o Paraganglioma?
O Feocromocitoma é o tipo mais comum de Tumor Neuroendócrino. Ele tem origem a partir das células cromafins, presentes na glândula suprarrenal e em outros gânglios do sistema nervoso simpático.
Essas células são produtoras de adrenalina, um hormônio que prepara o corpo para uma resposta de “luta ou fuga”. No sistema cardiovascular, a adrenalina provoca vasoconstrição e um aumento na frequência cardíaca, aumentando a oxigenação cerebral e muscular. Assim, o aumento da pressão arterial é uma das principais características do feocromocitoma.
Cerca de 80-85% dos tumores com origem nas células cromafins crescem na medula adrenal (camada interna da glândula adrenal), sendo denominados de Feocromocitomas. Os outros 15 a 20% crescem nos gânglios fora da glândula adrenal e são denominados de Paraganglioma.
A maioria desses tumores ficam restritos à glândula adrenal ou gânglios simpáticos. No entanto, cerca de 10% deles podem fazer metástases e se espalhar pelo corpo. Infelizmente, no momento, ainda não é possível dizer com base nas característica celular, se um feocromocitoma terá comportamento benigno (sem es espalhar) ou maligno (espelhando-se pelo corpo).
A maioria dos casos ocorre em pessoas com idade entre 30 e 50 anos de idade, ainda que muitas pessoas nunca venham a ser diagnosticadas.

Feocromocitomas X paragangliomas
O feocromocitoma e o paraganglioma são tumores neuroendócrinos raros com origens celulares semelhantes. Eles compartilham dos mesmos sinais e sintomas, mas diferem-se principalmente pela localização: o feocromocitoma forma-se na medula da glândula adrenal, enquanto os paragangliomas estão localizados em gânglios extra-adrenais.
Outras diferenças a serem consideradas incluem:
- Malignidade: Paragangliomas têm maior risco de serem malignos (metastáticos).
- Produção Hormonal: Ambos produzem catecolaminas (adrenalina, noradrenalina), mas paragangliomas podem secretar menos catecolaminas ou apenas dopamina, tornando-os, às vezes, não funcionais.
- Genética: Paragangliomas estão mais frequentemente associados a síndromes hereditárias.
Sinais e sintomas do feocromocitoma
Os feocromocitomas e paragangliomas podem ser quentes (produtores de hormônio) ou frios (não produtores de hormônios).
Paragangliomas localizados em gânglios na cabeça e pescoço geralmente são frios. Nesses casos, os sintomas gralmente estão associados ao efeito de massa, pelo crescimento do tumor – que pode se tornar doloroso e pode ser palpável sob a pele.
Já o feocromocitoma e demais paragangliomas, na maior parte das vezes serão quentes, produtores de hormônio. Nesses casos, os sintomas estão associados à liberação de adrenalina na corrente sanguínea.
A principal manifestação clínica é uma hipertensão arterial grave.e de difícil controle. Essa hipertensão está associada também a uma tríade de sintomas, que inclui cefaleia (dor de cabeça), sudorese e palpitação (taquicardia) – embora nem sempre os três simtomas estejam presentes.
Embora o feocromocitoma esteja longe de ser a principal causa para uma hipertensão arterial descontrolada, é preciso aventar esta possibilidade no caso de pessoas que precisam de dois ou três medicamentos anti-hipertensivos diferentes.
Cerca de 50% dos pacientes sintomáticos desenvolvem uma condição denominada de ataques paroxísticos. Esses pacientes apresentam uma liberação súbita de adrenalina, produzindo sintomas intensos e repentinos.
A pressão arterial pode subir para níveis bastante elevados, de até 25/20mmHg. Nestes níveis de hipertenssão, o paciente fica em risco elevado inclusive de complicações como o Acidente Vascular Cerebral ou o Infarto Agudo do Miocárdio.
Diagnóstico do feocromocitoma
Em caso de suspeita clínica de feocromocitoma, é feita a dosagem laboratorial da Metanefrina, um sub-produto da epinefrina. Isso pode ser feito por meio de um exame de urina de 24 horas ou por meio de um exame de sangue.
Níveis hormonais normais praticamente excluem o diagnóstico do feocromocitoma. Já um aumento de 2 a 3 vezes os valores de referência são bastante sugestivos, enquanto aumento maior do que 3 vezes dos valores de referência praticamente fecham o diagnóstico.
O diagnóstico por imagem deve ser feito apenas após a confirmação laboratorial, preferencialmente com a tomografia computadorizada do abdome com contraste. Quando a tomografia não identifica o tumor, o PET-CT deve ser solicitado para buscar identificar tumor extra-adrenal.
Cirurgia
Devido ao potencial para malignização do tumor, o tratamento do feocromocitoma é feito por meio de cirurgia, com a remoção da glândula adrenal (adrenalectomia).
Para isso, o paciente precisa de um acompanhamento multidisciplinar, incluindo o endocrinologista e o urologista.
Além deles, o anestesista e a equipe de terapia intensiva tem um papel muito importante no período perioperatório. Isso porque, ao mexer no tumor, as variações na pressão arterial podem sofrer oscilações significativas que exigem intervenção específica e imediata.
Para realizar a cirurgia, é fundamental que a hipertensão arterial esteja muito bem controlada.
Este controle geralmente leva de duas a seis semanas e envolve uma combinação de medicamentos anti-hipertensivos, especialmente os bloqueadores alfa e os betabloqueadores.
A cirurgia geralmente é feita por laparoscopia. Para isso, são utilizadas pequenas incisões para a introdução de um equipamento que contém uma câmera de vídeo em sua extremidade e também para a introdução dos instrumentos que auxiliarão na remoção do tumor.
Após a cirurgia, é fundamental que o controle rigoroso da pressão arterial seja mantido, o que exige um período de internação em Unidade de Terapia Intensiva (UTI).
A preocupação agora é o oposto de antes da cirurgia, ou seja, com uma queda muito rápida da pressão arterial devido à falta da adrenalina.
Isso pode exigir a aplicação de adrenalina por algumas horas ou dias, até que a pressão volte ao normal.
Aconselhamento genético
Cerca de 30% a 40% dos feocromocitomas estão associados a síndromes hereditárias, com herança autossômica dominante. A probabilidade de predisposição genética é ainda maior no caso de tumores bilaterais, metastáticos, multifocais, recorrentes ou de início precoce.
O aconselhamento genético é crucial em todos esses pacientes, uma vez que o conhecimento de mutações específicas ajuda a prever o comportamento do tumor, como o risco de malignidade, metástases ou recorrência, orientando um acompanhamento médico mais rigoroso. Além disso, ele ajuda na escolha do melhor tratamento e na vigilância de outras manifestações tumorais associadas às alterações genéticas.
O teste genético geralmente envolve painéis que investigam múltiplos genes, incluindo:
- VHL: Doença de von Hippel-Lindau.
- RET: Neoplasia Endócrina Múltipla tipo 2 (NEM2).
- NF1: Neurofibromatose tipo 1.
- SDHx (SDHA, SDHB, SDHC, SDHD): Síndromes de paraganglioma familiar.
- MAX, TMEM127.
Caso uma dessas variantes seja identificada, é importante que familiares de primeiro grau também realizem o teste genético. Caso esses familiares também sejam
portadores da mesma mutação, eles deverão realizar rastreamento bioquímico (metanefrinas plasmáticas ou urinárias) e de imagem (Ressonância/Tomografia) ao longo de toda a vida.
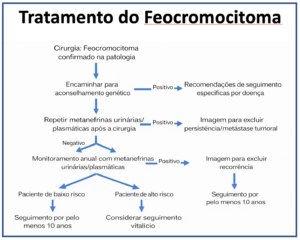
Monitoramento pós operatório
Apesar de um bom prognóstico geral, a doença pode recidivar em até 16% dos pacientes dentro de 10 anos após a cirurgia, com casos de recorrência às vezes em um longo prazo de acompanhamento.
Doença extra-adrenal, feocromocitomas hereditários, tumores do lado direito, tumores bilaterais e tumores maiores do que 3cm são considerados fatores de risco para recidiva. Além disso, até 30% dos feocromocitomas apresentam caracerísticas genéticas de risco, que podem inclusive ser transmitidas de pai para filho.
A ferramenta mais importante para o monitoramento pós-operatório é a dosagem de metanefrinas no sangue ou na urina de 24 horas. O aumento nos níveis de metanefrinas indica o retorno da produção de catecolaminas, o que sugere a persistência ou recorrência do tumor.
Caso as metanefrinas permaneçam elevadas após a cirurgia, exames de imagem devem ser realizados para avaliar a eventual persistência do tumor ou a presença de metástases.
Se as metanefrinas estiverem baixas, os exames deverão ser repetidos a cada 6 a 12 meses (a depender do risco do paciente), o que deve ser feito por pelo menos 10 anos (em pacientes de baixo risco) ou por toda a vida (nos pacientes de alto risco).