Feocromocitoma e Paraganglioma
O que é o feocromocitoma e o Paraganglioma?
O Feocromocitoma e o Paraganglioma são tumores neuroendócrinos raros que podem causar crises súbitas de aumento da pressão arterial, frequentemente acompanhadas de dor de cabeça intensa, palpitações e suor excessivo.
Esses tumores produzem quantidades excessivas de catecolaminas, como adrenalina e noradrenalina, responsáveis por ativar o sistema cardiovascular e desencadear os sintomas característicos.
Em muitos casos, os sintomas aparecem em “crises”, com períodos de melhora entre os episódios. As crises costumam surgir de forma inesperada, durar minutos ou horas e podem se repetir ao longo do tempo. Outras pessoas poderão ter hipertensão arterial persistente ou de difícil controle, mesmo com o uso de medicações.
Embora apresentem origem e características semelhantes, o feocromocitoma e o paraganglioma se diferenciam quanto a localização: o feocromocitoma forma-se na medula da glândula adrenal, enquanto os paragangliomas estão localizados em gânglios extra-adrenais.
O tratamento geralmente envolve a remoção cirúrgica do tumor, com controle cuidadoso da pressão arterial antes, durante e após o procedimento.

Sinais e sintomas do feocromocitoma
Pacientes com feocromocitoma ou paragangliomas podem ou não produzir hormônios catecolaminas. Quando produzem hormônios, a liberação pode ser contínua ou em pulsos. Os sintomas da doença dependem justamente dessas características, podendo os pacientes ser divididos em três grupos:
- Paragangliomas frios, não produtores de hormônios (10% dos casos)
- Paragangliomas quentes e com sintomas contínuos (60% dos casos)
- Paraganglionas quentes com crises paroxísticas (30% dos casos)
Paragangliomas frios
Os feocromocitomas e paragangliomas podem ser quentes (produtores de hormônio) ou frios (não produtores de hormônios).
Paragangliomas localizados em gânglios na cabeça e pescoço geralmente são frios. Nesses casos, os sintomas geralmente estão associados ao efeito de massa, pelo crescimento do tumor – que pode se tornar doloroso e pode ser palpável sob a pele.
Paragangliomas quentes e sintomas contínuos
Em cerca de 60% dos pacientes, a adrenalina é liberada de forma contínua. A principal característica clínica nesses casos é a hipertensão arterial refratária, que não atinge a meta pressórica mesmo com o uso de três ou mais medicamentos, incluindo um diurético.
Paragangliomas quentes com crises paroxísticas
Em cerca de 30% dos pacientes, as catecolaminas são liberadas em picos e sem um fator desencadeante claro. Esses pacientes desenvolvem sintomas em crises, denominadas de crises paroxísticas.
A tríade clássica é composta por três sintomas que costumam ocorrer juntos durante as crises:
- Cefaleia (dor de cabeça): Geralmente intensa, de início súbito e muitas vezes descrita como pulsátil. Pode durar minutos a horas e costuma acompanhar os picos de pressão arterial.
- Palpitações: Sensação de batimentos cardíacos acelerados ou irregulares. Reflete o efeito das catecolaminas (como adrenalina) sobre o coração.
- Sudorese: Episódios de sudorese profusa, muitas vezes generalizada, que surgem de forma abrupta durante as crises.
Esses três sintomas costumam ocorrer em conjunto durante episódios paroxísticos (crises), frequentemente acompanhados de aumento súbito da pressão arterial e sintomas como tremores, ansiedade ou sensação de pânico.
As crises podem durar de minutos a horas e se repetir de forma irregular, com períodos assintomáticos entre elas.
Quando suspeitar do feocromocitoma?
Quando suspeitar do feocromossitoma ou paraganglioma?
Clinicamente, devemos suspeitar do feocromocitoma especialmente em duas situações:
- Todo paciente que apresente hipertensão arterial de difícil controle e sem outra causa clara para isso, especialmente me pacientes mais jovens.
- Pacientes com crises recorrentes de cefaleia intensa, palpitações e sudorese (tríade clássica das crises paroxísticas).
Diagnóstico do feocromocitoma
Em caso de suspeita clínica, o primeiro passo é a dosagem laboratorial da Metanefrina, um sub-produto da epinefrina. Isso pode ser feito por meio de um exame de urina de 24 horas ou por meio de um exame de sangue.
Níveis hormonais normais praticamente excluem o diagnóstico. Já um aumento de 2 a 3 vezes os valores de referência são bastante sugestivos, e o aumento maior do que 3 vezes dos valores de referência praticamente confirma o diagnóstico.
Exames de imagem deve ser feito apenas após a confirmação laboratorial, preferencialmente com a tomografia computadorizada do abdome com contraste. Quando a tomografia não identifica o tumor, o PET-CT deve ser solicitado para buscar identificar tumor extra-adrenal.
Cirurgia
Devido ao potencial para malignização do tumor, o tratamento do feocromocitoma é feito por meio de cirurgia, com a remoção da glândula adrenal (adrenalectomia).
Para isso, o paciente precisa de um acompanhamento multidisciplinar, incluindo o endocrinologista e o urologista.
Além deles, o anestesista e a equipe de terapia intensiva tem um papel muito importante no período perioperatório. Isso porque, ao mexer no tumor, as variações na pressão arterial podem sofrer oscilações significativas que exigem intervenção específica e imediata.
Para realizar a cirurgia, é fundamental que a hipertensão arterial esteja muito bem controlada.
Este controle geralmente leva de duas a seis semanas e envolve uma combinação de medicamentos anti-hipertensivos, especialmente os bloqueadores alfa e os betabloqueadores.
A cirurgia geralmente é feita por laparoscopia. Para isso, são utilizadas pequenas incisões para a introdução de um equipamento que contém uma câmera de vídeo em sua extremidade e também para a introdução dos instrumentos que auxiliarão na remoção do tumor.
Após a cirurgia, é fundamental que o controle rigoroso da pressão arterial seja mantido, o que exige um período de internação em Unidade de Terapia Intensiva (UTI).
A preocupação agora é o oposto de antes da cirurgia, ou seja, com uma queda muito rápida da pressão arterial devido à falta da adrenalina.
Isso pode exigir a aplicação de adrenalina por algumas horas ou dias, até que a pressão volte ao normal.
Aconselhamento genético
Cerca de 30% a 40% dos feocromocitomas estão associados a síndromes hereditárias, com herança autossômica dominante. A probabilidade de predisposição genética é ainda maior no caso de tumores bilaterais, metastáticos, multifocais, recorrentes ou de início precoce.
O aconselhamento genético é crucial em todos esses pacientes, uma vez que o conhecimento de mutações específicas ajuda a prever o comportamento do tumor, como o risco de malignidade, metástases ou recorrência, orientando um acompanhamento médico mais rigoroso. Além disso, ele ajuda na escolha do melhor tratamento e na vigilância de outras manifestações tumorais associadas às alterações genéticas.
O teste genético geralmente envolve painéis que investigam múltiplos genes, incluindo:
- VHL: Doença de von Hippel-Lindau.
- RET: Neoplasia Endócrina Múltipla tipo 2 (NEM2).
- NF1: Neurofibromatose tipo 1.
- SDHx (SDHA, SDHB, SDHC, SDHD): Síndromes de paraganglioma familiar.
- MAX, TMEM127.
Caso uma dessas variantes seja identificada, é importante que familiares de primeiro grau também realizem o teste genético. Caso esses familiares também sejam
portadores da mesma mutação, eles deverão realizar rastreamento bioquímico (metanefrinas plasmáticas ou urinárias) e de imagem (Ressonância/Tomografia) ao longo de toda a vida.
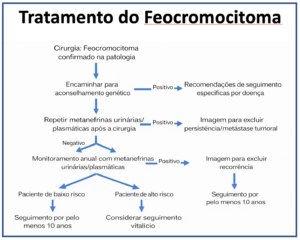
Monitoramento pós operatório
Apesar de um bom prognóstico geral, a doença pode recidivar em até 16% dos pacientes dentro de 10 anos após a cirurgia, com casos de recorrência às vezes em um longo prazo de acompanhamento.
Doença extra-adrenal, feocromocitomas hereditários, tumores do lado direito, tumores bilaterais e tumores maiores do que 3cm são considerados fatores de risco para recidiva. Além disso, até 30% dos feocromocitomas apresentam caracerísticas genéticas de risco, que podem inclusive ser transmitidas de pai para filho.
A ferramenta mais importante para o monitoramento pós-operatório é a dosagem de metanefrinas no sangue ou na urina de 24 horas. O aumento nos níveis de metanefrinas indica o retorno da produção de catecolaminas, o que sugere a persistência ou recorrência do tumor.
Caso as metanefrinas permaneçam elevadas após a cirurgia, exames de imagem devem ser realizados para avaliar a eventual persistência do tumor ou a presença de metástases.
Se as metanefrinas estiverem baixas, os exames deverão ser repetidos a cada 6 a 12 meses (a depender do risco do paciente), o que deve ser feito por pelo menos 10 anos (em pacientes de baixo risco) ou por toda a vida (nos pacientes de alto risco).