Síndrome do QT Longo
O que é a síndrome do QT longo?
A síndrome do QT longo é uma condição que afeta a atividade elétrica do coração, levando a um atraso na recuperação dos batimentos cardíacos entre um impulso e outro. Esse atraso, identificado no eletrocardiograma como um intervalo QT prolongado, pode aumentar o risco de arritmias cardíacas perigosas, inclusive com risco de parada cardíaca e morte.
Os sintomas podem variar bastante. A maioria dos pacientes são assintomáticos e nem mesmo sabem ser portadores da Síndrome do QT longo, ou apresentam episódios leves e ocasionais de palpitação. Eventualmente, o QT longo pode ser identificado em um exame de rotina nesses pacientes.
Outros pacientes podem apresentar diferentes formas de taquiarritmias em episódios recorrentes, que retornam em poucos segundos para o ritmo cardíaco normal. Esses episódios geralmente são desencadeados por esforço físico, emoções intensas ou uso de determinados medicamentos e podem ser percebidos por sintomas como palpitação, mal-estar e tontura.
Em alguns casos, a taquiarritmia pode ter uma duração maior (geralmente acima de 15 segundos), podendo provocar desmaio ou movimentos involuntários que lembram uma crise convulsiva.
Uma das formas mais preocupantes de taquiarritmia é chamada de Torsades de Pointes. Embora a maioria delas também se recupere sozinha, eventualmente ela pode evoluir para uma fibrilação ventricular, que é uma arritmia grave e potencialmente fatal.
O tratamento depende da causa e do risco individual, podendo incluir a suspensão de medicamentos desencadeantes, correção de alterações eletrolíticas, uso de medicamentos como betabloqueadores e, em casos selecionados, dispositivos como o cardiodesfibrilador implantável (CDI).
A Síndrome do QT longo é grave?
A Síndrome do QT longo é uma condição potencialmente grave. Embora aproximadamente 50% das pessoas nunca apresentem sintomas, em cerca de 10% dos casos a parada cardíaca é o primeiro sinal da doença.
Como a Síndrome do QT longo acomete o coração?
Para compreender o que acontece com o coração no paciente com a Síndrome do QT longo, é preciso antes entender como funciona o sistema elétrico em um coração normal.
Normalmente, os impulsos elétricos que fazem o coração contrair têm origem em uma estrutura chamada de nó sinulsal, localizado no átrio direito. É de lá que ele se espalha para os átrios (câmeras superiores do coração) e então para os ventrículos (câmeras inferiores do coração).
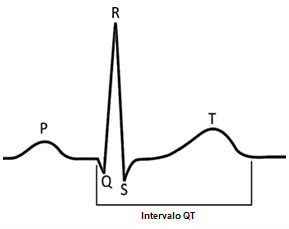
Ao receber o estímulo elétrico, as células do miocárdio (músculo do coração) se despolarizam e se contraem. A seguir, precisa se repolarizar, preparando-se para receber um novo estímulo.
No paciente com síndrome do QT longo essa repolarização é mais lenta do que o normal. O nome se refere às alterações que ocorrem em um eletrocardiograma – a onda QRS se refere à despolarização do ventrículo, enquanto a onda T se refere a sua repolarização.
Quais as causas da Síndrome do QT longo?
Algumas pessoas nascem com certas anormalidades genéticas que provocam um intervalo QT longo. Esses distúrbios envolvem defeitos nos canais iónicos presentes nas membranas celulares, especialmente nos músculos, coração e sistema nervoso. Por esse motivo, são também chamados de “canalopatias”. Quando presentes, esses defeitos fazem com que a repolarização das células seja mais lenta do que o normal.
Em outras pessoas, o intervalo QT longo não é hereditário, mas resulta de diferentes condições médicas, como baixos níveis séricos de potássio ou um ritmo cardíaco muito lento. Pode também estar associado ao uso de certos tipos de medicamentos, especialmente os antiarrítimos. Outros medicamentos que podem levar a um intervalo QT longo incluem certos antidepressivos, diuréticos, certos antivirais, antifúngicos ou antibióticos.
Quais os sintomas da Síndrome do QT longo?
Os sintomas podem ser bastante variáveis. Cerca de 50% dos pacientes nunca apresentarão sintomas e muitoas permancerão a vida toda sem saber ser portador da doença.
Quando presentes, os sintomas estão relacionados a episódios de taquiarritmias. Esses episódios duram poucos segundos e podem provocar sintomas como palpitação, mal-estar e tontura.
Em alguns casos, a taquiarritmia pode ter uma duração maior (geralmente acima de 15 segundos), podendo provocar desmaio ou movimentos involuntários que lembram uma crise convulsiva.
Eventualmente, a taquiarritmia pode evoluir para fibrilação ventricular, uma forma de parada cardíaca. Infelizmente, em cerca de 10% dos casos a parada cardíaca é o primeiro sinal da doença.
Diagnóstico
O diagnóstico da Síndrome do QT longo é feito a partir de um eletrocardiograma. Esse eletrocardiograma pode ser feito como parte de uma avaliação médica de rotina, quando feito por algum outro motivo não relacionado ao QT longo ou na avaliação de um paciente pós desmaio ou convulsão.
Uma vez que se identifique um paciente com a Síndrome do QT longo, todos os parentes de primeiro grau (irmãos, irmãs, pais e filhos) devem fazer um eletrocardiograma, bem como outros familiares com histórico de convulsões ou desmaios. Eventualmente a Síndrome do QT longo pode ser identificada nesses familiares, mesmo que assintomáticos e aparentemente saudáveis.
Nem todas as pessoas com a Síndrome do QT longo irá apresentar o registro de um intervalo QT longo em um ECG de rotina, já que essas alterações no exame são esporádicas, não estando presentes o tempo todo. O Hotter é um dispositivo que monitora o ritmo cardíaco ao longo de 24 horas, aumentando assim a probabilidade de encontro dos achados eletrocardiográficos característicos. O Teste de esforço cardiopulmonar (Teste Ergométrico) também pode ajudar para isso, já que o esforço físico pode favorescer o desenvolvimento da arritmia nesses pacientes.
Por fim, alguns casos em que o eletrocardiograma deonstra alterações limítrofes, o teste genético pode confirmar o diagnóstico.
Teste genético
O teste genético para a Síndrome do QT Longo (SQTL) geralmente é feito por amostra de sangue ou saliva (swab bucal). Ele identifica mutações no DNA) para confirmar o diagnóstico, avaliar riscos de arritmias fatais e guiar o tratamento.
O teste identifica uma mutação compatível em cerca de 75-80% dos casos, sendo que três genes principais (KCNQ1, KCNH2 e SCN5A) são responsáveis por cerca de 90% dos testes positivos. No entanto, um resultado negativo não exclui totalmente a síndrome, já que nem todas as mutações são conhecidas ou detectadas.
Uma vez identificada a mutação no paciente, familiares de primeiro grau devem ser testados para identificar se possuem a mesma mutação. Isso ajuda a adotar medidas preventivas apropriadas.
Diferentes tipos genéticos na Síndrome do QT longo
A Síndrome do QT Longo não é uma doença única, mas sim um conjunto de distúrbios genéticos que afetam os canais de íon do coração (canalopatias).
Até o momento, foram identificados mais de 15 subtipos diferentes (LQT1 a LQT15), mas cerca de 90% dos casos estão concentrados nos três primeiros (LQT1, LQT2 ou LQT3).
Cada uma dessas condições afeta os canais iônicos de uma forma diferente, o que tem relação com os gatilhos da arritmia e fatores de risco.
LQT1 (45% dos pacientes)
O LQT1 está ssociado a uma mutação no gene KCNQ1. Ela afeta os canais de potássio que deveriam retirar a carga positiva do coração para ele relaxar. A arritmia é geralmente desencadeada por esforço físico.
LQT2 (Aproximadamente 35%)
O LQT2 está associado a uma mutação no gene KCNH2. Ele também afeta canais de potássio, mas de uma forma diferente. Os gatilhos para attimia incluem emoções súbitas, sustos ou ruídos auditivos bruscos (despertadores, toque de telefone, latidos).
LQT3 (Aproximadamente 10%)
O LQT3 está associado a uma mutação no gene SCN5A. Nesses pacientes, os canais de sódio ficam “vazando” carga para dentro do coração durante o repouso. As arritmias geralmente acontecem durante o sono ou repouso absoluto. Embora menos comum que os dois tipos anteriores, os eventos arrítmicos no LQT3 geralmente são mais letais.
Tratamento
O tratamento da síndrome do QT longo adquirida consiste em substituir ou parar o uso de medicamentos que possam estar gerando a síndrome, além de corrigir eventuais distúrbios nutricionais ou metabólicos.
Quando não se tem uma causa clara para o problema, o tratamento é feito com medicamentos beta bloqueadoes. Os betabloqueadores controlam os sintomas em cerca de 70 a 80% dos pacientes. Nos casos que não respondem aos medicamentos, a cirurgia pode ser indicada.
A cirurgia é feita por meio da simpatectomia. Este procedimento envolve a remoção de nervos específicos do sistema nervoso simpático, o que ajuda a reduzir o risco de arritmia.
Nos casos em que falharem o tratamento com medicamentos e a cirurgia for contra-indicada, pode-se considerar o uso de marcapasso cardíaco (dispositivo que ajuda a regular o ritmo cardíaco) ou implante de um cardioversor desfibrilador implantável (CDI). O CDI detecta arritmias potencialmente fatais e aplica choques cardíacos automaticamente para prevenir morte súbita.
Medidas de Estilo de Vida
Certas medidas de estilo de vida podem reduzir o risco de desmaios ou morte súbita cardíaca associada à Síndrome do QT longo.
- Controle as emoções: irritação, estresse ou sustos podem desencadear a arritmia a cardíacos em algumas pessoas com síndrome do QT longo. Isso deve ser orientado para colegas e familiares. Deve-se também evitar quando possível ambientes propícios a esses sentimentos.
- Verificar os medicamentos: todo médico deve ser avisado sobre a síndrome, uma vez que certos medicamentos podem levar a um aumento no intervalo QT. Isso inclui também alguns medicamentos que podem ser comprados sem receita médica. É sempre válido também fazer uma “dupla checagem” antes de iniciar um novo medicamento.
- Atividades físicas: Praticar mais exercícios e praticar diferentes técnicas de mindfulness é uma forma de se conectar com outras pessoas e reduzir o estresse. No entanto, a atividade física pode também aumentar o risco de arritmia, de forma que isso deve ser feito sempre sob orientação do cardiologista. Discutimos mais sobre isso abaixo.
Atividade física
Para a maior parte dos pacientes, atividades físicas e esportivas não competitivas são recomendas. No esporte competitivo, as recomendações são atualmente um pouco menos restritivas do que já foram no passado, mas sempre baseada em uma decisão compartilhada entre a equipe médica e o paciente ou seus familiares.
Ainda que pessoas com QT longo tenham risco mais elevado do que a população em geral para morte súbita, estudos mostram necessidade relativamente baixa de resgate com desfibrilador externo, com três resgates com DEA em 1.700 pacientes-ano.
Isso é válido especialmente naquelas pessoas que apresentam alterações genéticas da síndrome, mas sem as manifestações clínicas ou eletrocardiográficas do QT longo.
Para atletas sintomáticos ou que apresentem as alterações características no eletrocardiograma, a participação em esportes competitivos pode ser considerada após o tratamento ter sido implementado e medidas de precaução apropriadas tomadas, assumindo que o atleta esteja assintomático e em tratamento por pelo menos 3 meses.
Uma exceção a isso são os esportes aquáticos. Pacientes com QT longo não devem entrar na água sozinhos e devem evitar a prática competitiva nessas modalidades, pelo risco de afogamento.
A orientação médica é sempre fundamental, uma vez que diferentes condições que levam a um intervalo QT longo podem ter diferentes prognósticos relacionados à atividade física.
Algumas recomendações gerais devem ser consideradas:
- Não realizar atividades aquáticas ou ao menos não fazer isso sozinho, pelo risco de afogamento.
- evitar a desidratação, exaustão pelo calor ou insolação
- estabelecer um plano de ação de emergência com a escola ou a equipe.
- Evitar condições que levem a uma carga emocional muito intensa, já que sentimentos como sustos ou irritação podem desencadear a arritmia.